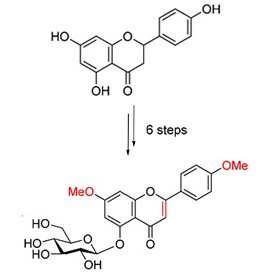
摘要:7,4'-二甲氧基洋芹素-5-O-葡萄糖苷作为珍贵中药白木香的有效成分具有抑制LPS 诱导巨噬细胞生成NO 活性.由于5-位羟基具有较强的分子内氢键, 5-位氧苷的黄酮类化合物在传统的糖苷化条件不能高效合成. 以价廉易得的柚皮素和D-葡萄糖为原料, 经选择性羟基保护、硼氢化钠还原、相转移催化下的糖苷化、2,3-二氯-5,6-二氰基对苯醌(DDQ)氧化等6 步反应, 以36.0%的总收率完成了7,4'-二甲氧基洋芹素-5-O-葡萄糖苷的化学合成, 为该化合物进一步的生物活性研究奠定了物质基础.
白木香[Aquilaria sinensis (Lour.) Gilg]为瑞香科(Thymelaeaceae)沉香属常绿乔木, 别名土沉香、女儿香、莞香等. 白木香为我国特有的珍贵药源植物, 野生资源曾经十分丰富, 但长期以来由于森林资源、生态环境遭受自然灾害和人为破坏等诸多原因, 白木香野生资源量在不断减少, 现仅分布有零星散生的残存植株, 1999 年白木香被国务院批准为国家二级重点保护野生植物, 现已载入《中国植物红皮书》.白木香产生的香脂即沉香, 是传统名贵药材和天然香料, 味辛、苦, 性微温, 沉香具有行气止痛、温中止呕、纳气平喘等功效. 用于治疗胸腹胀闷疼痛、胃寒呕吐呃逆、肾虚气逆喘急有显效; 在临床上用于治疗呃逆、功能性消化不良、痛经、前列腺痛、胃痛、胆汁反流性胃炎等. 中成药中以沉香组方配伍的有约200 种, 在《本草纲目》、《本草经疏》、《大明本草》以及《本草通玄》等药典书籍中均有记载. 现代医学试验研究表明, 白木香具有一定的抗肿瘤[1a~1c]、抗菌[2a~2b]、镇痛抗炎[3a~3b]、利泄[4]、降糖[5]等功能.5-位氧苷的黄酮、黄酮醇以及花青素类化合物是一类重要的天然产物, 它们广泛存在于自然界中[6a ~ 6f].7,4'-二甲氧基洋芹素-5-O-葡萄糖苷是北京大学屠鹏飞课题组首次从白木香茎木中分离得到的5-位氧苷黄酮化合物, 生物活性测试表明其具有抑制LPS 诱导巨噬细胞生成NO 活性, 其IC50 值为9.19 μmol•L-1, 明显优于阳性对照药布洛芬(IC50=94.12 μmol•L-1)[7]. 由于7,4'-二甲氧基洋芹素-5-O-葡萄糖苷在植物中含量较少, 分离提纯困难(原始文献从40 kg 的白木香茎木中仅分离纯化得到23 mg 的目标化合物), 因此限制了其在生物活性及作用机制等方面的深入研究. 经查阅文献, 目前尚未有该化合物的合成报道. 首次实现了7,4'-二甲氧基洋芹素-5-O-葡萄糖苷的化学合成, 为进一步研究其药理活性奠定物质基础, 并为化学合成其他5-位氧苷的黄酮及黄酮醇类化合物及其衍生物提供参考.
1 结果与讨论
由于分子内氢键的存在, 黄酮或黄酮醇类化合物中5-位羟基糖苷键的构建一直是合成中的一个难点问题.截止目前, 采用化学合成的方法完成黄酮或黄酮醇类化合物5-位氧苷的合成仅有少量的文献报道[8a~8f], 可分为直接法和间接法两种合成策略. 直接法的合成策略是指糖基供体与黄酮或黄酮醇母核的5-位酚羟基在催化剂的存在下发生糖苷化反应来直接构建5-位氧苷[8a~8e];间接法的合成策略则是糖基供体与黄烷母核的5-位酚羟基在催化剂的存在下发生糖苷化反应得到黄烷5-位氧苷化合物, 然后将黄烷5-位氧苷化合物经氧化后得到黄酮5-位氧苷化合物[8f]. 本文选择利用间接合成的策略对7,4'-二甲氧基洋芹素-5-O-葡萄糖苷进行合成.7,4'-二甲氧基洋芹素-5-O-葡萄糖苷的逆合成分析如下(图1): 目标化合物的制备可通过其前体黄烷糖苷6经DDQ 氧化成黄酮糖苷后脱除糖环上的保护基实现.黄烷糖苷6 可通过关键中间体5 与糖基供体8 发生糖苷化反应制备得到. 糖基供体8 可通过葡萄糖经全苯甲酰基保护后端基溴代而得, 而关键中间体5 则可由价廉易得的柚皮素2 为起始原料, 通过官能团转化制备得到.基于上述反合成分析, 以价廉易得的柚皮素2 为起始原料(图2), 经硫酸二甲酯选择性甲基化以86%的收率得到7,4'-二甲氧基柚皮素3; 化合物3 的5-位羟基经乙酰基保护, 使用95%乙醇重结晶, 以96%的收率得到白色固体化合物4[8f]; 二氢黄酮化合物4 中的4-位羰基经硼氢化钠还原, 后处理用乙酸乙酯-石油醚重结晶以91%的收率得到关键中间体黄烷化合物5[8f]. 在此需要指出的是, 曾尝试以5-位羟基裸露的化合物3 为原料进行4-位羰基的硼氢化钠还原, 结果反应体系变的较为复杂, 没有得到我们期望的关键中间体黄烷化合物5. 这可能是由于分子内氢键造成的.在得到关键中间体黄烷化合物5 后, 本路线的另一个关键问题即为黄烷化合物5-位氧苷键的构建. Midori小组[8f]对于黄烷化合物5-位氧苷键的构建采用氟代糖为糖基供体, 三氟化硼乙醚-2,6-二叔丁基-4-甲基吡啶(DTBMP)体系为促进剂. 采用他们的合成策略除了得到需要的5-位氧苷黄烷化合物以外, 还得到其重排的6-位碳苷黄烷化合物, 且两者之间难以实现分离纯化. 虽经过条件优化可以使重排产物所占比例降低, 但最终未能实现只得到唯一的5-位氧苷黄烷化合物的目标. 由此推测Midori 小组采用酸性的三氟化硼乙醚为糖苷化的促进剂是引起5-位氧苷黄烷化合物重排得到6-位碳苷黄烷化合物的一个重要原因.
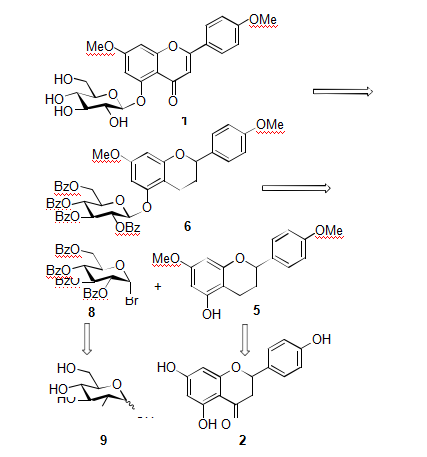

图1 7,4'-二甲氧基洋芹素-5-O-葡萄糖苷的逆合成分析
Figure 1 Retrosynthetic analysis of 7,4'-dimethylapigenin-5-O-glycoside
为克服Midori 小组的合成策略所存在的弊端, 采用溴代糖为糖基供体在碱性条件下来实现黄烷化合物5-位氧苷键的构建. D-葡萄糖全苯甲酰化后处理, 使用95%乙醇重结晶以84%的收率得到白色固体化合物10;全苯甲酰化葡萄糖10 采用乙酰溴-甲醇体系将端基溴代以88%的收率得到糖基供体8. 实验表明, 以全苯甲酰化的溴代葡萄糖作为糖基供体优于全乙酰化的溴代葡萄糖. 前者室温放置数月仍未见明显分解, 而后者密闭冷藏一周左右即会出现明显的颜色变化且伴有较多杂质的生成.在成功制备全苯甲酰化的溴代葡萄糖糖基供体8后, 首先试验了相转移催化下的糖苷化反应. 在相转移催化剂四丁基溴化铵存在的条件下, 将黄烷化合物5 与糖基供体8 首先室温搅拌下溶于氯仿, 然后向反应体系中加入等体积的0.5 mol/L 的碳酸钾溶液[9]. 令人高兴的是45 ℃下加热反应12 h 后以38%的收率得到5-位氧苷化合物6, 且反应中未检测到重排产物6-位碳苷化合物的生成. 当采用饱和碳酸钾溶液时, 45 ℃下加热反应18 h后能以66%的纯化后收率得到目标化合物6. 此外, 我们也曾尝试加热条件下, 黄烷化合物5 与糖基供体8 在N,N-二甲基甲酰胺(DMF)溶液中固体碳酸钾催化下的糖苷化反应, 但遗憾的是没有得到我们期望的目标产物[8d].


图2 7,4'-二甲氧基洋芹素-5-O-葡萄糖苷的合成路线
Figure 2 Synthesis of 7,4'-dimethylapigenin-5-O-glycoside
在DDQ 存在的条件下, 黄烷糖苷化合物6 可以被成功地氧化以78%的收率得到黄酮糖苷化合物7[8f]. 黄酮糖苷化合物7 在四氢呋喃-甲醇-水混合溶剂体系中[10], 采用2 equiv.的无水碳酸钾可以非常顺利地脱除糖环上的保护基以93%的收率得到需要的最终产物. 经过比对, 采用此法合成的7,4'-二甲氧基洋芹素-5-O-葡萄糖苷核磁共振图谱与天然产物数据一致[7].
2 结论
以价廉易得的柚皮素和D-葡萄糖为原料, 经选择性羟基保护、硼氢化钠还原、相转移催化下的糖苷化、DDQ 氧化等6 步反应, 以36.0%的总收率首次完成了7,4'-二甲氧基洋芹素-5-O-葡萄糖苷的化学合成. 终产物及中间体经NMR 和HRMS 结构确证. 该合成方法较之从白木香中提取7,4'-二甲氧基洋芹素-5-O-葡萄糖苷具有原料易得、操作简单、可大量制备等优点, 为后期深入研究其药理活性及结构改造奠定了物质基础. 同时本研究结果进一步丰富了5-位氧苷黄酮及黄酮醇类化合物及其衍生物的合成方法.
3 实验部分
3.1 仪器与试剂
熔点采用上海精密科学仪器有限公司制造WRR 型熔点仪测定; 核磁共振波谱采用Bruker VANCE-400 或
600 型核磁共振仪测定; 质谱采用Agilent 1946BESIMS型质谱仪测定; 旋光采用SGW-3 自动旋光仪测定.薄层色谱(TLC)采用HSGF254(烟台化学工业研究所), 柱层析使用硅胶为200~300 目(烟台化学工业研究所). 所用试剂和溶剂为市售化学纯或分析纯.
3.2 实验方法
3.2.1 2-(4-甲氧基苯基)-5-羟基-7-甲氧基苯并二氢吡喃-4-酮(3)的合成
将柚皮素2 (5.00 g, 18.38 mmol)溶于丙酮150 mL,室温搅拌下依次加入无水碳酸钾(10.14 g, 73.48 mmol)及硫酸二甲酯(7.04 mL, 73.56 mmol). 室温搅拌10 min后, 50 ℃下加热回流48 h, 经薄层色谱(TLC) [V(石油醚)∶V(乙酸乙酯)=10∶1]检测显示反应完全. 过滤,滤饼用丙酮洗涤, 减压蒸除大部分的溶剂后将剩余液倾入1 mol/L 盐酸200 mL 中, 二氯甲烷萃取(100 mL×2),合并有机相, 饱和食盐水100 mL 洗涤, 无水硫酸钠干燥. 减压浓缩柱层析得白色固体3 4.75 g, 收率86%.m.p. 119~120 ℃; 1H NMR (400 MHz, CDCl3) δ: 12.04(s, 1H), 7.38 (d, J=8.1 Hz, 2H), 6.95 (d, J=8.2 Hz, 2H),6.06 (s, 1H), 6.04 (s, 1H), 5.36 (d, J=12.9 Hz, 1H), 3.83(s, 3H), 3.80 (s, 3H), 3.21~3.00 (m, 1H), 2.78 (d, J=17.1Hz, 1H); 13C NMR (150 MHz, CDCl3) δ: 196.16, 168.08,164.25, 163.02, 160.17, 130.51, 127.86, 114.35, 103.25,95.20, 94.34, 79.12, 55.79, 55.49, 43.30; HRMS calcd for C17H15O5 299.0920, found 299.0986.
3.2.2 2-(4-甲氧基苯基)-5-乙酰氧基-7-甲氧基苯并二氢吡喃-4-酮(4)的合成
将化合物3 (1.67 g, 5.56 mmol)溶于二氯甲烷75mL, 室温搅拌下依次加入三乙胺(6.15 mL, 44.48 mmol)及DMAP (0.68 g, 5.56 mmol). 向反应体系中缓慢滴加乙酰氯(0.96 mL, 13.90 mmol), 10 min 后, 经TLC [V(石油醚)∶V(乙酸乙酯)=6∶1]检测显示反应完全. 将反应液倾入1 mol/L 盐酸200 mL 中, 二氯甲烷萃取(100 mL×2), 合并有机相, 饱和食盐水100 mL 洗涤,无水硫酸钠干燥. 减压浓缩得粗品, 粗品经95%乙醇重结晶, 过滤, 干燥得白色固体4 1.82 g, 收率96%. m.p.160~161 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.37 (d, J=8.3 Hz, 2H), 6.95 (d, J=8.4 Hz, 2H), 6.41 (s, 1H), 6.28 (s,1H), 5.40 (d, J=13.6 Hz, 1H), 3.83 (s, 3H), 3.82 (s, 3H),3.04 (t, J=16.4 Hz, 1H), 2.70 (d, J=16.7 Hz, 1H), 2.39 (s,3H); 13C NMR (150 MHz, CDCl3) δ: 189.22, 169.71,165.59, 164.47, 160.20, 151.98, 130.56, 127.91, 114.36,108.07, 104.84, 99.65, 79.46, 55.94, 55.51, 44.97, 21.27;HRMS calcd for C19H18NaO6 365.1001, found 365.1021.
3.2.3 2-(4-甲氧基苯基)-5-羟基-7-甲氧基苯并二氢吡喃(5)的合成
将化合物4 (1.37 g, 4.00 mmol)悬浮于30 mL 四氢呋喃及15 mL 水的混合溶剂中, 冰浴搅拌下少量多次加入硼氢化钠(0.91 g, 24.00 mmol), 3.5 h 后经TLC [V(石油醚)∶V(乙酸乙酯)=6∶1]检测显示反应完全. 向反应体系中加入饱和氯化铵终止反应, 乙酸乙酯萃取(100 mL×2), 合并有机相, 饱和食盐水100 mL 洗涤,无水硫酸钠干燥. 减压浓缩得粗品, 粗品经石油醚-乙酸乙酯重结晶, 过滤, 干燥得白色固体5 1.04 g, 收率91%. m.p. 137~139 ℃; 1H NMR (400 MHz, CDCl3) δ:7.35 (d, J=8.5 Hz, 2H), 6.92 (d, J=8.5 Hz, 2H), 6.12 (d,J=2.1 Hz, 1H), 6.01 (d, J=2.1 Hz, 1H), 5.02~4.85 (m,2H), 3.83 (s, 3H), 3.73 (s, 3H), 2.79~2.61 (m, 2H), 2.26~2.14 (m, 1H), 2.12~1.97 (m, 1H); 13C NMR (150 MHz,CDCl3) δ: 159.48, 159.36, 157.03, 154.65, 133.74, 127.59,114.09, 101.96, 94.81, 94.48, 55.48, 55.44, 29.37, 19.26;HRMS calcd for C17H18O4 286.1205, found 286.1187.
3.2.4 5-(β-2,3,4,6-四苯甲酸酯-D-葡萄糖氧基)-2,3-二氢-2-(4-甲氧基苯基)-7-甲氧基苯并二氢吡喃(6)的合成
将化合物5 (0.77 g, 2.69 mmol), 化合物8 (2.67 g,4.05 mmol)溶于氯仿50 mL, 室温搅拌下依次加入四丁基溴化铵(0.44 g, 1.36 mmol)及饱和碳酸钾溶液50 mL.室温搅拌10 min 后, 45 ℃下加热反应18 h, 经TLC[V(石油醚)∶V(乙酸乙酯)=4∶1]检测显示反应完全.
分液, 水相用二氯甲烷萃取(40 mL×2), 合并有机相,饱和食盐水50 mL 洗涤, 无水硫酸钠干燥. 减压浓缩柱层析得白色固体6 1.53 g, 收率66%. m.p. 198~199 ℃;[α]25D -27.8 (c 0.38, CHCl3); 1H NMR (400 MHz, CDCl3)
δ: 8.05 (d, J=7.7 Hz, 2H), 7.98~7.93 (m, 4H), 7.87 (d,J=7.7 Hz, 2H), 7.57~7.23 (m, 14H), 6.91~6.83 (m, 2H),6.35 (s, 1H), 6.18 (s, 1H), 6.05~5.96 (m, 1H), 5.85 (t, J=8.7 Hz, 1H), 5.73 (t, J=8.2 Hz, 1H), 5.39 (t, J=7.5 Hz,1H), 4.83~4.65 (m, 2H), 4.56~4.44 (m, 1H), 4.36 (s,1H), 3.80 (s, 3H), 3.64 (s, 3H), 2.75 (d, J=16.8 Hz, 0.5H),2.65~2.53 (m, 0.5H), 2.45 (d, J=13.1 Hz, 0.5H), 2.35~2.21 (m, 0.5H), 1.91 (s, 1.5H), 1.75~1.66 (m, 0.5H); 13CNMR (150 MHz, CDCl3) δ: 166.32, 165.91, 165.40,165.27, 165.17, 159.47, 159.44, 159.30, 156.78, 156.70,155.94, 155.83, 133.72, 133.66, 133.60, 133.54, 133.50,133.25, 130.04, 129.95, 129.86, 129.62, 129.30, 129.23,128.86, 128.78, 128.62, 128.50, 127.56, 127.51, 114.03,105.13, 99.78, 99.57, 96.13, 95.63, 95.57, 77.73, 72.88,72.82, 71.70, 71.64, 69.66, 63.44, 55.45, 29.10, 19.57,19.40; HRMS calcd for C51H45O13 865.2860, found865.2847.
3.2.5 5-O-(β-2,3,4,6-四苯甲酸酯-D-葡萄糖)-7,4'-二甲氧基洋芹素(7)的合成
将化合物6 (0.66 g, 0.77 mmol)溶于二氯甲烷60mL, 室温搅拌下加入DDQ (0.53 g, 2.31 mmol). 18 h 后,经TLC [V(石油醚)∶V(乙酸乙酯)=4∶1]检测显示反应完全. 过滤, 二氯甲烷洗涤滤饼, 有机相用饱和食盐水50 mL 洗涤, 无水硫酸钠干燥. 减压浓缩柱层析得淡黄色固体7 0.53 g, 收率78%. m.p. 131~132 ℃; [α]25D-32.8 (c 0.35, CHCl3); 1H NMR (400 MHz, CDCl3) δ:8.03~7.89 (m, 8H), 7.74 (d, J=8.6 Hz, 2H), 7.54~7.29(m, 12H), 6.98 (d, J=8.6 Hz, 2H), 6.71 (s, 1H), 6.58 (s,1H), 6.39 (s, 1H), 6.07~5.89 (m, 2H), 5.81 (t, J=9.3 Hz,1H), 5.66 (d, J=6.8 Hz, 1H), 4.68 (d, J=11.7 Hz, 1H),4.49 (dd, J=11.9, 5.7 Hz, 1H), 4.32 (s, 1H), 3.87 (s, 3H),3.77 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 176.31,166.21, 165.99, 165.36, 163.25, 162.16, 160.69, 159.17,156.69, 133.61, 133.36, 133.13, 132.97, 130.13, 130.05,129.94, 128.91, 128.57, 128.45, 128.39, 128.29, 127.72,123.98, 114.46, 110.61, 107.56, 103.59, 99.84, 96.50,73.07, 72.93, 72.82, 69.62, 63.23, 55.85, 55.60; HRMScalcd for C51H41O14 877.2496, found 877.2487.
3.2.6 7,4'-二甲氧基洋芹素-5-O-葡萄糖苷(1)的合成
将化合物7 (287 mg, 0.33 mmol)溶于甲醇5 mL、四氢呋喃5 mL 以及水1 mL 的混合溶剂中, 室温搅拌下加入无水碳酸钾(90 mg, 0.66 mmol). 室温搅拌反应5 min后, 加热至45 ℃继续反应2 h, 经TLC [V(石油醚)∶V(乙酸乙酯)=1∶4]检测显示反应完全. 加入阳离子树脂中和至中性, 过滤, 甲醇洗涤,有机相减压浓缩得粗品, 将所得粗品悬浮于10 mL 乙酸乙酯, 加热回流30min 后, 自然冷却, 过滤, 滤饼用乙酸乙酯洗涤, 真空干燥得黄色固体1 140 mg, 收率93%. m.p. 210~211 ℃;[α] 25D - 40.5 (c 0.45, pyridine); 1H NMR (400 MHz,DMSO-d6) δ: 8.04 (d, J=7.9 Hz, 2H, H-2', 6'), 7.11 (d, J=7.9 Hz, 2H, H-3',5'), 7.10 (s, H-8, 1H), 6.91 (s, H-6, 1H),6.81 (s, H-3, 1H), 4.76 (d, J=6.6 Hz, H-1'', 1H), 3.90 (s,7-OCH3, 3H), 3.86 (s, 3H, 4'-OCH3), 3.75 (dd, J=12.0,5.0 Hz, 1H, H-6''), 3.52(m, H-6'', 1H), 3.50~3.16 (m, 4H,H-2'', 3'', 4'', 5''); 13C NMR (150 MHz, DMSO-d6) δ:176.95 (C-4), 163.61 (C-7), 162.10 (C-4'), 160.96 (C-2),158.48 (C-9), 158.24 (C-5), 128.04 (C-2'), 122.75 (C-1'),114.55 (C-3'), 109.30 (C-10), 106.49 (C-3), 104.18 (C-1''),103.53 (C-6), 96.61 (C-8), 77.66 (C-3''), 75.77 (C-5''),73.61 (C-2''), 69.95 (C-4''), 60.95 (C-6''), 56.09 (C-7-OMe), 55.56 (C-4'-OMe); HRMS calcd for C23H25O10461.1448, found 461.1439.
3.2.7 1,2,3,4,6-五苯甲酸酯-D-葡萄糖(10)的合成
将D-葡萄糖9 (9.00 g, 50.00 mmol)溶于吡啶150mL, 冰浴下缓慢滴加苯甲酰氯(43.70 mL, 375.00 mmol),滴加完毕后撤去冰浴, 室温继续搅拌反应4 h, 经TLC[V(石油醚)∶V(乙酸乙酯)=2∶1]检测显示反应完全.加入甲醇10 mL 终止反应, 减压蒸除大部分溶剂后将剩余液倾入1 mol/L 盐酸200 mL 中, 二氯甲烷萃取(100mL×2), 合并有机相, 饱和食盐水100 mL 洗涤, 无水硫酸钠干燥.减压除去溶剂, 95%乙醇重结晶得白色粉末10 29.46 g, 收率84%.
3.2.8 1-溴-2,3,4,6-四苯甲酸酯-D-葡萄糖(8)的合成
将化合物10 (7.00 g, 10.00 mmol)溶于冰醋酸100mL, 室温氮气保护下依次加入乙酰溴 (3.70 mL, 50.00mmol)及甲醇(1.20 mL, 30.00 mmol), 室温搅拌8 h, 经TLC [(V 石油醚)∶V(乙酸乙酯)=6∶1]检测显示反应完全. 向反应体系中加入二氯甲烷300 mL 稀释, 分别用冷水、饱和碳酸氢钠水溶液、水各200 mL 洗涤, 无水硫酸钠干燥, 减压浓缩柱层析得白色固体8 5.81 g, 收率88%. m.p. 129~130 ℃; [α]25D -28.1 (c 0.45, CHCl3);1H NMR (400 MHz, CDCl3) δ: 8.07 (d, J=7.4 Hz, 2H),8.00 (d, J=7.4 Hz, 2H), 7.96 (d, J=7.6 Hz, 2H), 7.88 (d,J=7.5 Hz, 2H), 7.62~7.49 (m, 3H), 7.47~7.36 (m, 7H),7.33~7.29 (m, 2H), 6.87 (s, H-1, 1H), 6.27 (t, J=10.0 Hz,H-4, 1H), 5.82 (t, J=10.1 Hz, H-3, 1H), 5.33 (d, J=10.4Hz, H-2, 1H), 4.74~4.65 (m, 2H), 4.51 (d, J=12.3 Hz,1H); HRMS calcd for C41H33O11 701.2023, found701.2019.
链接文本:
http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract346782.shtml




![1-甲基-4-[[4-硝基-2-(三氟甲基)苯基]甲基]-哌嗪](http://struc.chem960.com/casimg/694500/vpvwpqyew1xymmqisubos63wee.png)